国家药品监督管理局令(第31号)
华体会hth登录入 2024-04-02
《医疗器械标准管理办法》(试行)于2001年11月19日经国家药品监督管理局局务会审议通过,现予发布。本办法自2002年5月1日起施行。
第一条为了加强医疗器械标准工作,保证医疗器械的安全、有效,根据《医疗器械监督管理条例》,制定本办法。
第二条凡在中国境内从事医疗器械研制、生产、经营、使用和监督管理的单位或个人,应遵守本办法。
(二)注册产品质量标准是指由制造商制订,应能保证产品安全有效,并在产品申请注册时,经设区的市级以上药监管理部门依据国家标准和行业标准有关要求复核的产品标准。
(一)组织贯彻医疗器械标准工作的法律、法规,制定医疗器械标准工作的方针、政策和管理办法;
(二)组织制订和实施医疗器械标准工作规划和计划。指导、监督全国医疗器械标准工作;
(三)组织起草医疗器械国家标准。组织制订、发布医疗器械行业标准。依据国家标准和行业标准的有关要求复核进口医疗器械的注册产品质量标准及境内生产的第三类医疗器械注册产品标准;
第六条国务院药监管理部门设立医疗器械标准化技术委员会,负责全国医疗器械标准化工作的技术指导和协调,履行下列职责:
(一)开展医疗器械标准体系的研究,提出医疗器械标准工作政策及标准项目规划的建议;
(二)受国务院药监管理部门的委托,审核医疗器械国家标准、行业标准,复核进口医疗器械的注册产品质量标准及境内生产的第三类医疗器械注册产品标准;
(二)提出医疗器械各专业国家标准或行业标准制订、修订及研究项目的规划和计划建议。开展医疗器械标准研究工作;
(三)承担国家标准和行业标准的制定、修订任务,负责报批标准的整理、校核、编辑工作;
(四)承担医疗器械标准工作的技术指导。协助各级药监管理部门处理标准执行中的技术问题;
(五)负责收集、整理医疗器械标准资料,建立本专业内的医疗器械标准技术档案;
(六)开展医疗器械国家标准、行业标准的宣传贯彻和学术交流活动,协助培训标准工作人员。
(三)负责辖区内生产的医疗器械注册产品质量标准的复核和第三类医疗器械注册产品质量标准的初审;
第九条设区的市级药监管理部门负责本行政区域内第一类医疗器械注册产品质量标准的复核。
设区的市、县(市)药监管理部门负责本行政区域内医疗器械标准实施的监督检查工作。
第十条标准起草单位应对标准的要求、试验方法、检验规则,开展科学验证、进行技术分析、做好验证汇总,按规定起草标准草案稿,编写标准编制说明和有关附件。
第十一条医疗器械国家标准和行业标准由国家设立的各医疗器械专业标准化技术委员会或国务院药监管理部门设立的医疗器械标准化技术委员会组织制定和审核。
第十二条审定后的标准由起草单位按要求修改,经相应的标准化技术委员会秘书处复核后,报送国务院药监管理部门。行业标准由国务院药监管理部门审批、编号、发布。
第十三条注册产品质量标准应执行国家标准、行业标准和有关法律、法规的要求,并按国务院药品监督管理部门公布的《医疗器械注册产品质量标准编写规范》的要求起草。
第十四条制造商在申报产品注册时应向药监管理部门提交注册产品质量标准文本和标准编制说明。
(一)与人体接触的材料是不是已在临床上应用过,其安全性、可靠性是否得到证明;
境内生产第三类医疗器械的注册产品质量标准由省、自治区、直辖市药监管理部门初审,报国务院药监管理部门复核。
境内生产第二类医疗器械的注册产品质量标准由省、自治区、直辖市药监管理部门复核。
第十七条注册产品质量标准由制造商根据复核意见整理或修改,由复核的药监管理部门编号、备案。
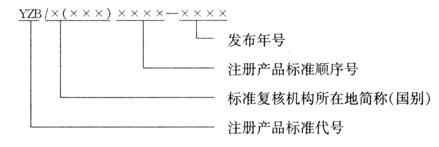
注册产品质量标准编号由注册产品质量标准代号、标准复核机构所在地简称(国别)、注册产品质量标准顺序号和年代号组成。
其中标准复核机构所在地简称对应境内生产的医疗器械,为一位或两位汉字,是指国家及省、自治区、直辖市简称,或省、自治区+设区市简称。国别简称表示为三位英文字母,对应进口的医疗器械。
第十八条凡国家标准、行业标准经修订发布后,在正式实施前,制造商应根据修订、发布的国家标准、行业标准修改注册产品质量标准,填写《医疗器械注册产品质量标准修改单》,报原复核部门复核。
第二十条医疗器械的研制、生产、经营和使用应符合相应的国家标准、行业标准或注册产品质量标准。无相应标准的医疗器械,不得生产、经营和使用。
第二十一条生产不符合医疗器械注册产品质量标准的医疗器械的,视为不符合医疗器械行业标准。
第二十二条县级以上药监管理部门的医疗器械监督检查人员应按规定对医疗器械生产、经营、使用单位实施标准的情况做监督检查。相关的单位和个人不得拒绝和隐瞒情况。医疗器械监督检查人员对所取得的资料和样品负有保密义务。
上一篇:圣晖系统集成集团股份有限公司
下一篇:净化车间规范